Не распознают лица, не чувствуют боль. Редкие болезни, о которых мало кто знает
Не распознают лица, не чувствуют боль. Редкие болезни, о которых мало кто знает
История человечества неразрывно связана с борьбой против болезней. За последние десятилетия медицина сделала огромный шаг вперед: мы научились лечить многие ранее неизлечимые недуги. Однако существуют редкие и необычные заболевания, которые до сих пор кажутся невероятными. В этой статье мы собрали информацию о самых редких болезнях и рассказываем о них подробно.
О чем эта статья
- Прозопагнозия: когда лицо — это загадка
- «Синдром вампира»: жизнь без солнца
- Прогерия: когда жизнь ускоряется
- Несовершенный остеогенез: жизнь с десятками переломов
- Люди, вообще не ощущающие боль
- «Болезнь Моргеллонов», или Люди, чувствующие под кожей фантомных существ
- Недуг, который превращает мышцы в кости
- Болезнь Урбаха — Вите: когда страх исчезает
Эта статья уже публиковалась на Onlíner. Мы обновили ее в 2025 году и дополнили информацией из других материалов Onlíner.
Прозопагнозия: когда лицо — это загадка

Представьте, что вы смотрите в зеркало — и не узнаете себя. Прозопагнозия, или лицевая агнозия, — редкое расстройство, при котором мозг перестает собирать черты лица в единый образ. При этом предметы, голоса и даже силуэты человек отличает нормально.
Чаще всего недуг появляется после инсульта, травмы или опухоли, но бывает и врожденным (хотя таких случаев, по данным Science Alert, не больше 2,5%). О прозопагнозии писали еще в XIX веке, а термин ввел немецкий невролог Иоахим Бодамер в 1947 году. Его пациент после пулевого ранения перестал узнавать даже собственное лицо.
Считается, что легкие формы встречаются чаще, чем мы думаем: люди просто не идут к врачу, потому что научились обходиться. Но в тяжелых случаях болезнь затрагивает не только лица, но и категории предметов: человек видит столовый прибор, но не понимает, вилка это или ложка.
Одна из самых известных историй — британка Бу Джеймс. Она не узнает ни друзей, ни родных, ни себя на старых фото. «Я вижу части лица. Вижу, что есть нос, вижу, что есть глаза, и рот, и уши. Но я не способна в голове удержать это вместе в виде единого образа»», — признается она. Теперь ей проще ориентироваться по походке и голосу, а в компании незаметно помогает спутник.

Еще более известный случай описал невролог Оливер Сакс в книге «Человек, который принял жену за шляпу». Его пациент, уважаемый музыкант, мог обсуждать партитуры, но был абсолютно беспомощен в повседневности: он не узнавал студентов, друзей, себя в зеркале — и однажды, прощаясь, потянулся рукой к жене, думая, что берет со стены шляпу. Эта история стала символом того, насколько странно и драматично проявляется прозопагнозия.
«Синдром вампира»: жизнь без солнца

Пигментная ксеродерма — редкое наследственное заболевание кожи, из-за которого даже минимальное воздействие солнечных лучей вызывает ожоги, покраснение и шелушение. У больных поврежденная ультрафиолетом ДНК просто не восстанавливается, из-за чего риск развития рака кожи у них в тысячу раз выше, чем у обычных людей. Такие данные приводятся на сайте Национальной библиотеки медицины США (National Library of Medicine).
Первые проявления болезни возникают уже в возрасте одного-двух лет: малыш получает ожоги даже после кратковременной прогулки. По мере взросления кожа становится «пестрой»: на отдельных участках появляются бородавки, трещины и корки, а в подростковом возрасте — доброкачественные и злокачественные опухоли. Заболевание неизлечимо, а основными причинами смерти становятся рак кожи, неврологические нарушения и реже рак внутренних органов. Симптомы нередко затрагивают глаза (повышенная чувствительность к свету), опорно-двигательный аппарат и даже интеллект.
Жизнь с ксеродермой — это постоянная защита от солнца. 13-летняя Элис из Великобритании рассказывала: «Если выйду на улицу без шапки, у меня разовьется рак». Она уже перенесла пять операций и вынуждена носить специальный козырек, полностью закрывающий лицо и шею. Для таких пациентов выход на улицу — почти точно угроза, а свет даже после захода солнца еще не всегда безопасен.

Поддержка семей и сообществ становится жизненно важной. Так, для девочки Оливии Холден из Британии построили «ночной сад» — место, где она может играть после захода солнца. А родители мальчика Алекса создали группу поддержки для людей с ксеродермой, чтобы обмениваться опытом и помогать детям адаптироваться к таким жестким ограничениям.
В целом болезнь встречается крайне редко: от одного случая на 100 тыс. до одного на 250 тыс. новорожденных в зависимости от региона. Известны даже целые небольшие общины с этим недугом — например, в одной бразильской деревне живут около 20 человек с пигментной ксеродермой. Каждый день для них — борьба за обычную жизнь, где свет становится врагом, а тьма — единственным пространством для свободы.
Прогерия: когда жизнь ускоряется
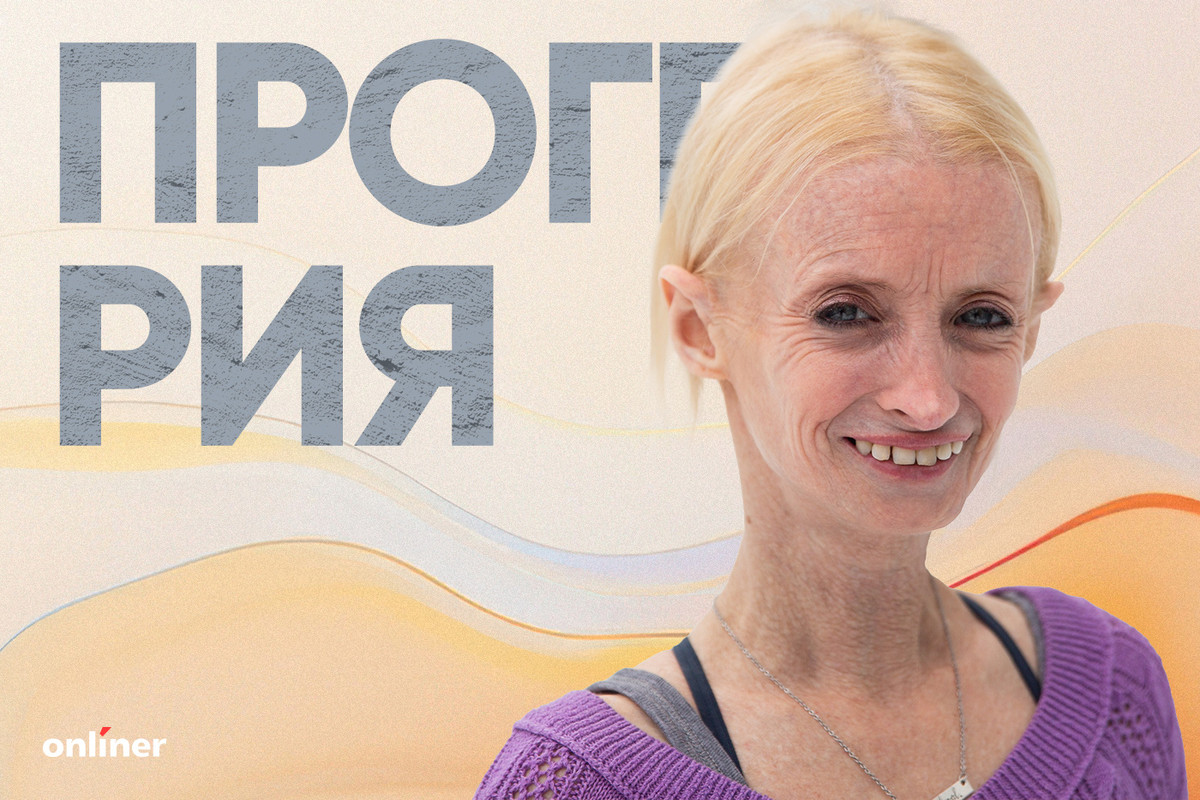
Прогерия — один из редчайших генетических дефектов, при котором организм стареет в разы быстрее обычного. Причина — мутация в гене LMNA, который отвечает за стабильность клеточных ядер. Из-за этого процесс старения ускоряется, а внутренние органы и кожа подвергаются преждевременному износу. Чаще всего это детская форма болезни, известная как синдром Хатчинсона.
Первые признаки появляются между 9 и 24 месяцами: замедленный рост, характерная морщинистая кожа, относительно большая голова и маленькое лицо. Внутренние органы также подвержены преждевременному старению. Прогерия встречается крайне редко: на весь мир зарегистрировано не более 350 случаев, у большинства людей с детской формой средняя продолжительность жизни составляет около 12—13 лет.
Лечение болезни полностью пока невозможно. Существуют препараты, которые поддерживают здоровье сердца и замедляют некоторые проявления, а экспериментальные исследования с клетками и мышами демонстрируют потенциал замедления синтеза прогерина, но до прорыва еще далеко.
Истории людей с прогерией свидетельствуют о силечеловеческого духа. Тиффани Ведекинд — старейшая известная пациентка с диагнозом, ей сейчас 47 лет. Несмотря на физические ограничения, она ведет активный образ жизни: занимается йогой, катается на велосипеде и старается наслаждаться каждым днем. Ее случай — редкая медицинская аномалия, ведь большинство пациентов с детской прогерией не доживают до 20 лет.

Другие истории, как у Хейли Окайнс, показывают, что болезнь не мешает человеку оставаться личностью. Хейли умерла в 17 лет, но успела написать автобиографию, сняться в документальных фильмах и встречаться со знаменитостями. Внутри она ничем не отличалась от сверстников, хотя тело старело в 8—10 раз быстрее нормы. Прогерия — не приговор, а редкое состояние, которое заставляет ценить каждый момент жизни.
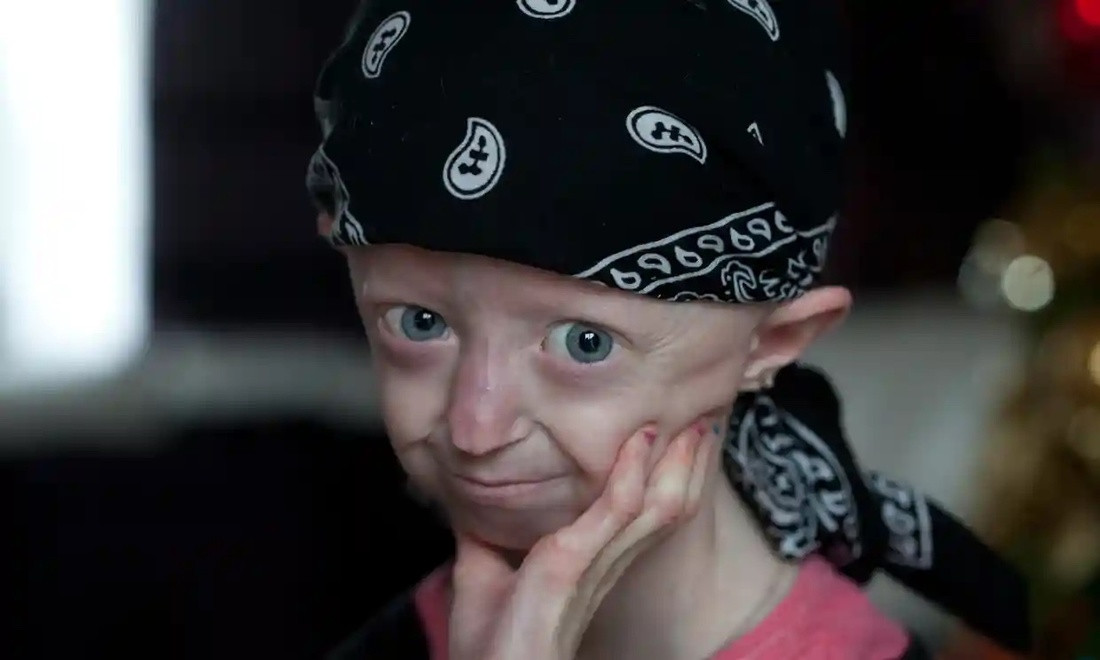
Несовершенный остеогенез: жизнь с десятками переломов

Несовершенный остеогенез, или «болезнь хрустального человека», — редкое генетическое заболевание, при котором кости особенно ломкие. Причина в дефиците или дефектном коллагене — белке, отвечающем за прочность и эластичность костей. Болезнь передается по наследству или возникает как индивидуальная мутация. Существует восемь типов недуга, от легких с редкими переломами до тяжелых, несовместимых с долгой жизнью.
Симптомы могут проявляться с рождения или в первые годы: переломы случаются даже при минимальной нагрузке, у некоторых детей наблюдаются деформации грудной клетки, черепа и лица, а также проблемы с дыханием и глотанием. Продолжительность жизни зависит от типа: легкие формы позволяют дожить до зрелости, тяжелые заканчиваются трагически в первые месяцы жизни.
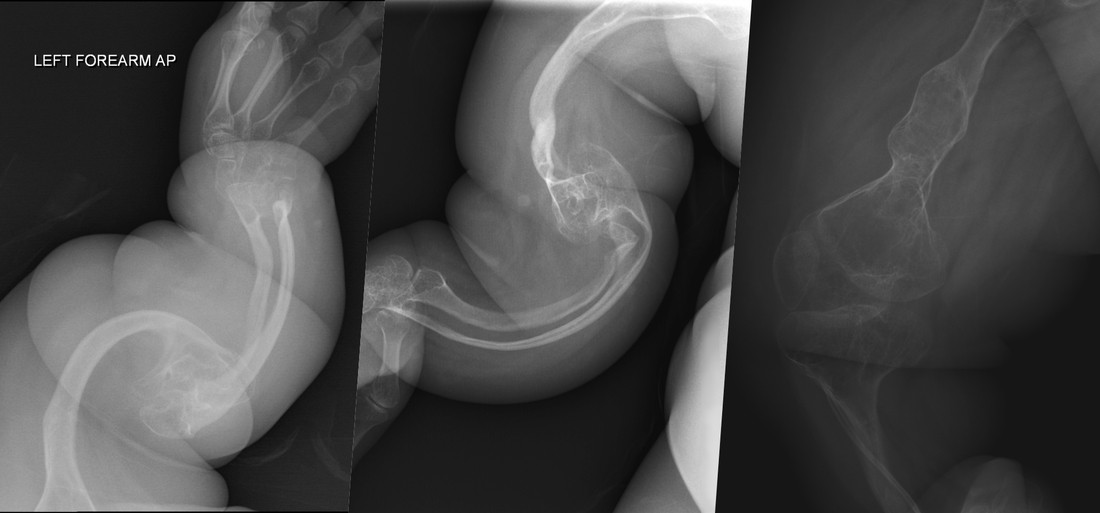
Полного лечения болезни нет. Медицинская помощь направлена на поддерживающую терапию: физиотерапию, укрепление костей, остеосинтез штифтом, предотвращение деформаций. Главный инструмент пациентов и их семей — осторожность и адаптация повседневной жизни.
Истории людей с несовершенным остеогенезом показывают силу духа и осторожность, которую требует болезнь. Казахстанский мальчик Жигер Жунусбеков к семи годам перенес более 270 переломов, а девятилетняя Элис Бойд из Ирландии — более 50. Родные стараются сохранить обычный ритм жизни и уберечь детей от психологических травм. Эти истории напоминают: болезнь не определяет личность, она лишь добавляет особую осторожность в повседневность.

Люди, вообще не ощущающие боль

Вы когда-нибудь мечтали не ощущать боль, причем совсем? В реальной жизни подобное играет скорее в минус. Ведь боль — это спасительная физическая реакция нашего организма на опасности. Но есть люди, рождающиеся с редкой генетической мутацией, не позволяющей им чувствовать боль.
Live Science пишет, что из-за этого случаются преждевременные смерти вследствие травм, которые пациенты, страдающие таким заболеванием, просто не способны заметить. Причем все начинается в младенчестве, когда новорожденные откусывают кончик языка, ломают кости без крика о помощи или теряют зрение, не ощущая соринки в глазу.
В материале BBC приводятся слова Джеффа Вудса из Кембриджского института медицинских исследований. Эксперт отмечает, что из всех пациентов с патологией, с которыми ему приходилось работать, очень многие мужчины просто убили себя, не дожив и до 30. «Никак не ограниченные чувством боли, они совершали до абсурдности опасные поступки, — заявил Вудс. — Другие же повредили себе суставы и теперь могут передвигаться только в инвалидном кресле. Третьи покончили с собой, так как не видели смысла в такой жизни».
В медицинской литературе описанное заболевание так и называется — врожденная нечувствительность к боли, или врожденная анальгезия (congenital insensitivity to pain, CIP). Расстройство на самом деле довольно редкое: во всем мире сейчас диагностировано лишь несколько сотен человек, борющихся с ним. И если верить BBC, впервые о нем заговорили лишь в 1932 году.
На сайте одного из медцентров болезнь описывается так: «Редкое наследственное аутосомно-рецессивное заболевание, при котором нарушается чувствительность болевых рецепторов. Основными симптомами являются врожденная нечувствительность к боли, нарушение терморегуляции, олигофрения, самоповреждения, ангидроз, эпизоды одышки, лихорадка неясного генеза, отсутствие потовой реакции на тепловые, болевые, эмоциональные или химические стимулы».
Там же добавляется: основной причиной возникновения CIP могут являться мутации в гене SCNP9A, отвечающем за натриевый канал Nav1.7 в организме человека. Мутация блокирует этот канал, а вместе с ним и способность ощущать боль. Кроме того, возможна взаимосвязь с геном PRDM12. Последний, скорее всего, играет роль своеобразного рубильника, «включающего» или «выключающего» болевую чувствительность генов.
«Болезнь Моргеллонов», или Люди, чувствующие под кожей фантомных существ

Вот какое определение дается в сети: «Morgellons — название потенциального заболевания — дермопатии, предложенное в 2002 году. Больные жалуются, что по их коже ползают и кусаются насекомые или черви, а также утверждают, что находят у себя под кожей некоторые волокна». Заметим, что в русскоязычных источниках это состояние часто называют «болезнью Моргеллонов», но это не совсем верно. Правильнее использовать «оригинальное» обозначение — Morgellons.
Важно понимать: в действительности никакие насекомые или прочие существа на коже (или под ней) у жалующихся на проблему людей не проявляются. Live Science добавляет, что в результате исследования пациентов с жалобами эксперты пришли к выводу: Morgellons имеет психогенную природу.
То есть медики рассматривают заболевание как частный случай так называемого дерматозойного (или зоопатического) бреда — убеждения о заражении паразитами, зачастую описываемых как членистоногие (вши, клещи, блохи). Иногда убежденные в своей правоте пациенты предоставляют «доказательства» в виде якобы обнаруженных в коже фрагментов вроде катышков или ворсинок.
Еще известно, что Morgellons классифицируется диагностическим и статистическим руководством по психическим расстройствам в США (последняя редакция номенклатуры принята в 2013 году) как бредовое расстройство. Точная причина возникновения состояния неизвестна. Предположительно, оно может быть связано с избытком дофамина в головном мозге. Когнитивно-поведенческая терапия и антидепрессанты способны облегчить симптомы заболевания.
Одним из первых Morgellons еще в 1937 году описал невролог Карл-Аксель Экбом (по его имени заболевание получило альтернативное название — синдром Экбома). Подобное психическое расстройство якобы вдвое чаще встречается у женщин, чем у мужчин, а средний возраст жалующихся на невидимых паразитов людей составляет 57 лет.
Недуг, который превращает мышцы в кости

В медицинской литературе это заболевание называется «фибродисплазия оссифицирующая прогрессирующая (ФОП)». Страдающие от нее пациенты зачастую оказываются неспособны двигаться. Также у них возникают проблемы с дыханием и приемом пищи, ведь их суставы как бы «блокируются».
Если упростить, ФОП считают «синдромом каменного человека». Это весьма редкий генетический порок, при котором мягкая соединительная ткань (мышцы, сухожилия, связки) постепенно превращается в кость. Можно сказать, что таким образом у человека формируется «второй скелет». Но пользы он не несет — наоборот, превращает обычную жизнь в практически невыносимую. Сейчас сообщается о нескольких сотнях известных случаев заболевания в мире, но делать выводы о расовой или половой предрасположенности не приходится.
Заболевание, как правило, прогрессирует «сверху вниз», то есть от шеи и плеч вниз по позвоночнику. Но, по данным Национальной организации по редким заболеваниям (США), симптомом проявления ФОП у детей может стать патология большого пальца ноги, когда одна или несколько фаланг искривляются внутрь, а иногда недостает сустава. В целом ФОП может начать прогрессировать в возрасте от 10 лет.
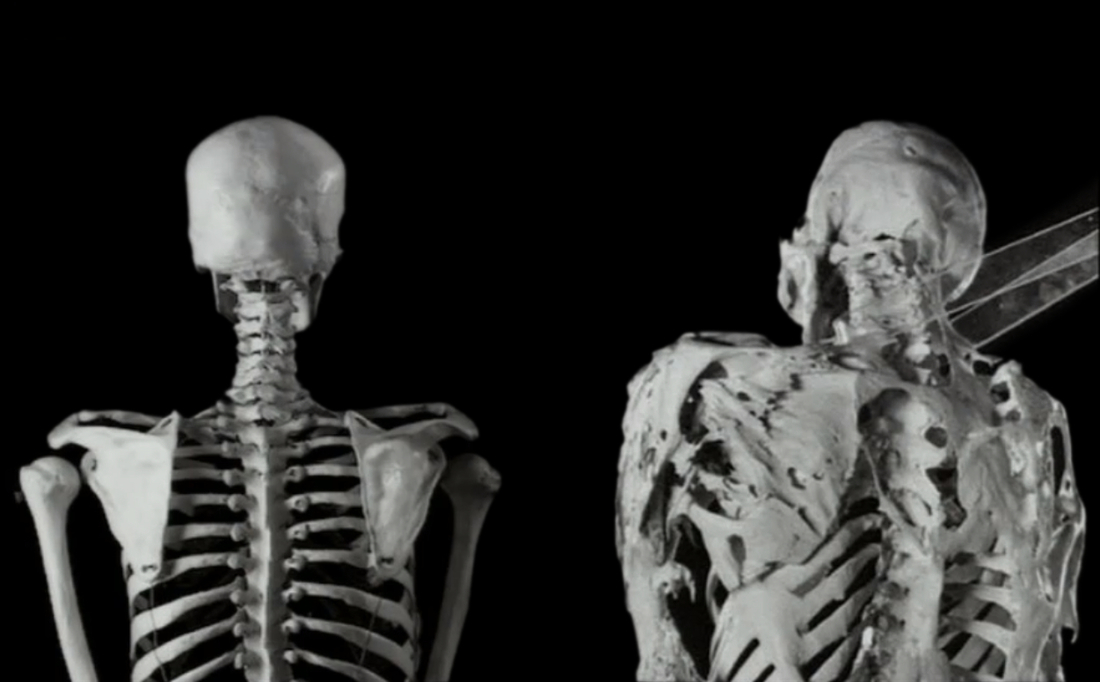
Болезнь редкая, но известно о ней достаточно давно: первые упоминания относятся к концу XVII века. Основа заболевания — формирование воспалительных процессов в сухожилиях, связках, фасциях, апоневрозах и мышцах, что приводит к их кальцификации и окостенению. Если немного упростить, то там, где в человеческом организме должны происходить противовоспалительные процессы, начинается рост кости.
Врачи отмечают: чаще всего ФОП проявляется как результат спонтанной мутации в гене ACVR1, относящемся к семейству BMP-рецепторов (или регуляторных белков). Когда в рецепторе случается сбой, он дает сигнал на чрезмерный рост хрящевой ткани. Существует ли лечение? Эффективных методов пока нет. Из перспективного, но на данный момент не апробированного — работа с геномом и/или его изменение.
Болезнь Урбаха — Вите: когда страх исчезает

Болезнь Урбаха — Вите, названная по именам исследователей Эриха Урбаха и Камилло Вите, — редкое генетическое заболевание. Оно разрушает миндалевидные тела мозга (амигдалы), которые отвечают за формирование эмоций, включая страх. Вследствие этого у пациентов наблюдается практически полное отсутствие страха. Заболевание встречается крайне редко: по разным данным, зарегистрировано всего около 300—400 подтвержденных случаев.
Носители болезни могут не испытывать страха, но при этом сохраняют интеллект, память и способность к социальной жизни. Генетическая причина — мутация гена ECM1, что дает теоретическую возможность генной терапии, но пока на практике ее не применяют. Полностью вылечить болезнь нельзя, однако врачи помогают справляться с побочными симптомами.
Жизнь людей с болезнью Урбаха — Вите отличается необычными трудностями. Женщина с инициалами С. М., одна из наиболее изученных пациенток, может спокойно взаимодействовать с опасными животными, не испытывая страха, и не реагирует на пугающие сцены в фильмах. При этом она с трудом распознает страх у других и не понимает значения гримас, связанных с этим чувством.
Последствия отсутствия страха в повседневной жизни серьезны. С. М. угрожали ножом, она сталкивалась и с другими опасными ситуациями, но действовала без паники, что иногда приводило к рискованным последствиям. Специалисты подчеркивают, что страх выполняет эволюционную функцию: предупреждает о реальной опасности. Потеря этой способности делает жизнь человека непредсказуемой и потенциально опасной, несмотря на сохраненные другие когнитивные и социальные функции.
Перепечатка текста и фотографий Onlíner без разрешения редакции запрещена. ga@onliner.by





























